Medicina Individualizada: a genética das doenças cardiovasculares hereditárias
Diagnóstico, prognóstico e aconselhamento familiar nos portadores de cardiomiopatias e canalopatias herdadas
Por Dra. Ilana Zalcberg*
Com o termo Doenças Cardiovasculares Hereditárias (DCH) nos referimos a um grupo de entidades que são caracterizadas por apresentação familiar causadas por raras variantes genéticas (mutações) com penetrância alta ou relativamente alta, sendo penetrância a proporção de indivíduos carregando uma variante genética que também expressam um fenótipo relacionado a uma doença específica.
Esse é um grupo de doenças cardíacas heterogêneas, que inclui as cardiomiopatias (doenças do músculo do miocárdio), canalopatias (doenças cardíacas pela deficiência de proteínas de “canal”, responsáveis por transmitir os impulsos elétricos do coração) e outras síndromes com envolvimento vascular caracterizadas pelo componente genético e de apresentação familiar.
As DCH apresentam uma série de características comuns que fazem com que o diagnóstico genético seja útil não só para individualizar o diagnóstico, mas também para personalizar o prognóstico e tratamento dessas doenças: são caracterizadas por sua componente genética e de apresentação familiar; sua apresentação clínica é altamente variável, em muitos casos com fenótipos sobrepostos, sendo difícil prever a progressão da doença; centenas de mutações diferentes têm sido relatadas em associação com estas doenças.
As DCH são uma das principais causas de morte súbita em jovens e atletas, mas também são uma causa significativa entre os pacientes mais velhos. A morte súbita pode ser a primeira manifestação dessas doenças – diante de um caso assim, os médicos consideram sempre o diagnóstico de cardiomiopatias e canalopatias. Tal situação indica a necessidade de um diagnóstico precoce, que permite a realização de uma avaliação cuidadosa do risco de morte súbita e a tomada de medidas preventivas eficazes.
As DCH são geralmente doenças monogênicas (uma variante genética única é responsável pela doença, em geral com herança mendeliana), mesmo quando uma proporção substancial de pacientes afetados carrega mais de uma variante patogênica. Nós incluímos nesse grupo as cardiomiopatias (Hipertrófica, di- latada, restritiva, arritmogénica e não compactada), canalopatias (QT longo e curto, Síndrome de Brugada, taquicardia ventricular polimórfica catecolaminérgica, distúrbios de condução herdados), assim como as doenças hereditárias afetando a aorta (Marfan, Loeys-Dietz, aneurisma familiar da aorta torácica).
Estas são condições clinicamente heterogêneas. As manifestações morfológicas e funcionais (como a presença de obstrução ou o grau e distribuição da hipertrofia na cardiomiopatia hipertrófica), associadas com manifestações extracardíacas (como miopatia esquelética em algumas cardiomiopatias ou perda de audição na SQTL), alterações eletrocardiográficas, idade de início, prognóstico e muitas outras características são altamente variáveis em cada uma das DCH. Essa heterogeneidade clínica é em parte explicada pela heterogeneidade genética subjacente, com vários genes diferentes e mutações associadas com uma doença e também múltiplas doenças associadas com o mesmo gene.
O DIAGNÓSTICO GENÉTICO
A genética tem sido o “padrão ouro” para a reavaliação dos critérios de diagnóstico para os parentes em cardiomiopatias hipertróficas e arritmogênicas. Entretanto, os testes genéticos não devem ser interpretados de forma isolada – devem ser submetidos à avaliação de um cardiologista em conjunto com um geneticista.
Devido ao conhecimento crescente da variabilidade genética nos genomas humanos, e à necessidade de diferenciar polimorfismos genéticos neutros das mutações associadas patogenicamente às doenças, é extremamente importante que qualquer variação descrita num paciente preencha os critérios para ser considerada uma mutação.
Uma mutação patogênica associada a uma CMH é uma alteração na sequência de DNA que não é observada ou é extremamente rara na população saudável e altera ou é preditiva em relação a uma alteração da estrutura e/ou função da proteína por ela codificada, ou foi associada conclusivamente à doença em uma família. Para isso, é importante que os resultados obtidos do sequenciamento de genes de um paciente sejam comparados com um banco de dados das mutações descritas antes de serem associados definitivamente ao fenótipo.
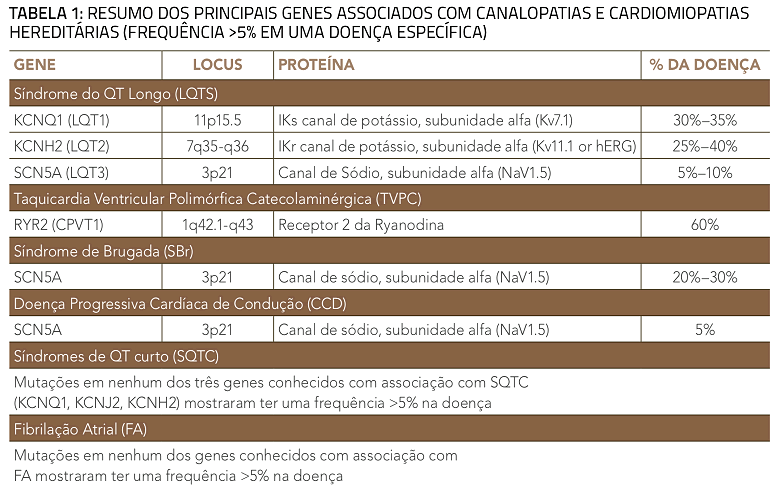
Os genes comumente associados à CMH e canalopatias, incluindo somente aqueles responsáveis por pelo menos 5% da frequência para uma determinada doença, são vistos na tabela 1. A frequência de detecção de mutações pelos testes genéticos é diferente de acordo com a doença, variando nas canalopatias a partir de 20% para a síndrome de QT curto e 75% para a síndrome do QT longo. Nas cardiomiopatias, as frequências variam entre 20% para a cardiomiopatia restritiva e 60% para cardiomiopatia hipertrófica familiar.
CARDIOGENÉTICA BASEADA EM EVIDÊNCIAS
A interpretação inadequada dos resultados dos estudos genéticos pode levar a decisões clínicas inapropriadas, assim como à utilização dos estudos genéticos em pacientes nos quais esses estudos não são indicados, gerando assim custos desnecessários. Para isso, o papel de normatização e orientação das sociedades científicas e grupos de estudo têm sido de fundamental importância. Entretanto, conhecimento e treinamento são uma necessidade para executar adequadamente as indicações e obter as interpretações dos testes genéticos.
A tabela 2 apresenta as recomendações de consenso da Heart Rhythm Society (HRS) e da European Heart Rhythm Association (EHRA) para uso dos estudos genéticos em canalopatias e miocardiopatias. Nesse consenso, a recomendação de Classe I (“Recomendado”) foi aplicada para o teste genético em casos-índice com uma suspeita clínica forte para a presença de uma canalopatia ou uma cardiomiopatia, quando o valor preditivo positivo de um teste genético é elevado (probabilidade de resultado positivo >40% e relação sinal/ ruído >10), e/ou quando o resultado do teste genético fornece informações tanto para diagnóstico ou prognóstico, como pode influenciar as escolhas terapêuticas. Uma recomendação de Classe IIa (“Pode ser útil”) ou Classe IIb (“Pode ser considerado”) foi aplicada quando alguma das condições anteriormente citadas não estava presente. A recomendação de Classe III (“Não recomendado”) foi aplicada nos casos em que o resultado do teste genético não forneceu nenhum benefício adicional ou poderia ser prejudicial na avaliação diagnóstica dos pacientes com possível doença cardíaca hereditária.
A triagem dos membros da família para a mutação identificada no probando foi recomendada como Classe I quando o teste genético leva à adoção de terapia / medidas cautelares / adaptações de estilo de vida. Inversamente, os autores atribuíram uma recomendação Classe IIa quando os resultados dos testes genéticos não estão associados com a utilização de medidas terapêuticas ou de proteção, mas os resultados podem ser úteis para o aconselhamento reprodutivo ou casos em que o teste genético é solicitado pelo paciente que quer saber o seu estado de mutação. Ao usar a orientação desse documento, é importante lembrar que não há uma regra absoluta que rege as diversas situações clínicas. Afinal, o julgamento sobre um determinado paciente deve ser feito pelo médico e pelo paciente à luz de todas as circunstâncias relevantes. Os autores do Consenso alcançaram um acordo igual ou superior a 84% em todas as recomendações; a maioria das recomendações teve concordância de 94% ou superior.
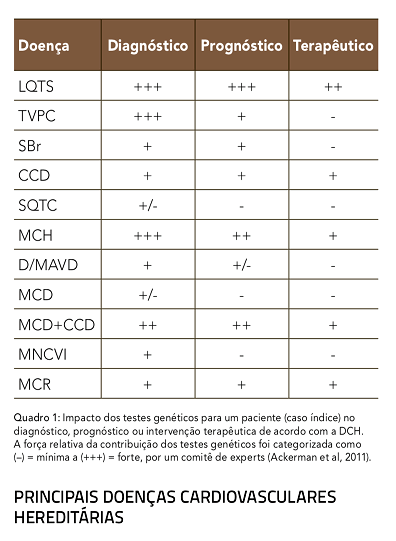
Para sua implementação correta na clínica, os testes genéticos devem ser avaliados na contribuição ou benefício trazido por estes para a tríade Diagnóstico – Prognóstico – Terapêutica, como apresentado no quadro 1



CARDIOMIOPATIA HIPERTRÓFICA (CMH): GENÉTICA COMO FATOR ESSENCIAL NO DIAGNÓSTICO DIFERENCIAL
A CMH é uma doença comum (afeta uma em cada 500 pessoas), caracterizada por hipertrofia cardíaca sem causa conhecida, alterações dos miócitos e fibrose. Muitos pacientes são assintomáticos e são diagnosticados na vida adulta durante check-ups rotineiros. Entretanto, a CMH pode causar invalidez e ser potencialmente letal devido às suas manifestações de dispneia, angina ao exercício, palpitações, síncopes e morte súbita.
De fato, a CMH é a causa mais comum de morte súbita em adultos jovens e atletas. A morte súbita pode ser a primeira manifestação da doença, por isso o diagnóstico precoce e uma avaliação de risco adequada são essenciais. O modo de herança é autossômica dominante e, portanto, a prole de um afetado tem uma chance de 50% de herdar a mutação causal. Subjacente à doença, foi descrito um grande número de mutações em genes diferentes; esta ampla variedade de causas genéticas corresponde à grande variabilidade na apresentação clínica, expressão morfológica, evolução e fatores prognósticos da doença. Esta é uma característica que a CMH compartilha com outras cardiomiopatias herdadas. A penetrância (ou seja, a proporção de indivíduos com mutação que têm doença clinicamente detectável) aumenta com a idade, mas permanece incompleta.
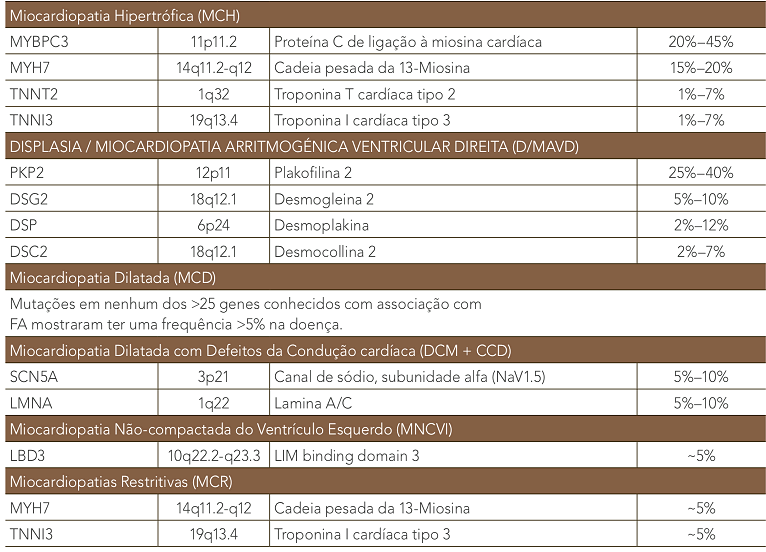
A CMH foi denominada “doença do sarcômero” quando os primeiros três genes identificados na doença mostraram codificar componentes do aparelho contráctil. Mutações em pelo menos dez genes de proteínas de sarcômero têm sido associadas causalmente à CMH. Os genes MYH7, codificando a cadeia pesada beta-miosina, e MYBPC3, codificando Proteína C de ligação à miosina cardíaca, são os mais comumente afetados por mutações patogênicas, cada um representando 25-35% de todos os casos, com cada um dos genes restantes contribuindo 1-5% (tabela 1). A detecção de mutações e o tipo destas são de extrema importância para o diagnóstico da doença. A CMH isolada é usualmente causada por mutações nos genes sarcoméricos e a ecocardiografia pode ser usada para guiar os testes genéticos. Dos dois subtipos morfológicos de MCH mais comuns, a MCH com curvatura septal reversa tem uma chance de 80% de um teste genético positivo, enquanto que apenas uma minoria de pacientes com MCH com septo ventricular sigmoidal terão um teste positivo.
Embora um teste negativo não possa afastar o diagnóstico de MCH, um teste positivo, além de firmar esse diagnóstico, tem um papel importante no diferencial entre CMH e cardiomegalia adaptativa ao exercício (“coração de atleta”), assim como com feno-cópias (Fabry, Noonan, LEOPARD, etc.) para as quais há abordagens clínico-terapêuticas diferenciadas.
Nestes casos, mutações em genes não sarcoméricos estão associadas às formas não isoladas da doença (sindrômicas, metabólicas, neuromusculares). Estas incluem a ocorrência da CMH no contexto clínico de uma síndrome genética (p.e. Síndrome de Noonan ou Costello); transtornos metabólicos do glicogênio (doença lisossomal de Pompe, Fa- bry, Danon, e na síndrome CMH/Wolff-Parkinson-White, HCM/WPW); defeitos nas cadeias respiratórias mitocondriais e doenças neuromusculares e neurodegenerativas como Duchenne/Becker e ataxia de Friedreich. A chance de identificar esses transtornos é maior na infância que na vida adulta e a CMH pode ser distinguida dessas doenças por exames clínicos e diagnóstico de DNA.
Além disso, a quimioprevenção pode ser possível no futuro próximo, em que estratégias clínicas e farmacológicas poderão ser usadas para prevenir ou atrasar o início da hipertrofia naqueles pacientes portadores de mutação, mas ainda sem manifestação clínica. Atualmente, um estudo randomizado está testando um tratamento profilático com diltiazem em pacientes com CMH com genótipo positivo / hipertrofia negativa.
SÍNDROME DO QT LONGO: O ESTUDO GENÉTICO NA TRÍADE DIAGNÓSTICO- PROGNÓSTICO-TERAPÊUTICA
Com uma prevalência de cerca de uma em 2.500 pessoas, as SQTLs compreendem um grupo distinto de canalopatias cardíacas, caracterizadas por uma repolarização cardíaca retardada que se manifesta como uma prolongação do QT no contexto de um coração de estrutura normal. Os pacientes têm um risco aumentado de síncopes, crises convulsivas e morte súbita, usualmente precipitada por estímulos físicos, emocionais ou auditivos, ou durante o período pós-parto. Como todas as cardiopatias apresentadas nesta revisão, as SQTLs são caracterizadas por penetrância incompleta e expressividade variável. As SQTLs são geneticamente heterogêneas e a forma mais comum de heranças é autossômica dominante.
Centenas de mutações têm sido identificadas nos 12 genes associados às SQTL, com aproximadamente 75% dos casos associados a mutações em 3 genes críticos que orquestram o potencial de ação dos miócitos ventriculares: KCNQ1 (LQT1), KCNH2 (LQT2) e SCN5A (LQT3) (tabela 1). O restante dos casos apresenta mutações em genes que codificam proteínas que participam em outros canais cardíacos. Estes genes, associados com SQTL4 a 12 contribuem com 5% das mutações.
Mais do que qualquer DCH, a associação Fenótipo-Genótipo pode orientar o estudo genético nas SQTLs. Por exemplo, eventos cardíacos induzidos pela natação ou outros exercícios indicam fortemente a presença de mutações no gene KCNQ1, enquanto que a indução via estímulos auditivos, assim como os eventos pós-parto, sugerem KCNH2 (LQT2), e eventos durante o sono são associados com SCN5A (LQT3). Em contraste, desmaios durante o exercício no contexto de um intervalo QT normal corrigido (QTc < 460 ms) deveria tirar a QTL1 do foco diagnóstico e sugerir TVPC.
Os estudos genéticos não somente têm implicações diagnósticas significativas, mas também têm impacto prognóstico e terapêutico. Por exemplo, a base genética subjacente influência fortemente a resposta ao tratamento padrão das SQTLs (bloqueadores beta), uma vez que a ação de b-bloqueadores é extremamente eficiente nos pacientes com SQTL1, mas não tanto nos pacientes com SQTL2 ou 3. Em contraste, alvejar o canal de sódio QTL3 patológico com agentes como mexiletina, flecainida, ranolazina ou propranolol pode representar uma opção terapêutica gene-dirigida na SQTL3.
Além disso, a estratificação de risco intrageno-típica (ou seja, baseada nas variações genéticas existentes nas mutações causadoras de um mesmo genótipo) tem sido útil nas SQTL 1 e SQTL2; nesses casos devem ser considerados o tipo e a localização da mutação no gene e na função celular. Por exemplo, pacientes com mutações “missense” (mudança de aminoácido) e localizadas no domínio transmembrana do gene LQT1 têm um risco aumentado em 2 vezes de um evento cardíaco quando comparados a pacientes com SQTL1 e mutações no domínio C-terminal do gene LQT1.
Pacientes com SQTL1 portadores de mutações que resultam em uma perda de função maior do canal de sódio (proteínas truncadas) têm um risco 2 vezes aumentado de evento cardíaco quando comparados a pacientes com a mutação no LQT1 que leva a um dano menor à proteína do canal.
Já pacientes com SQTL2 secundários a mutações na região gênica que codifica o poro do canal de potássio têm um QTc mais longo, manifestações clínicas mais severas e uma frequência maior de arritmias em idades mais precoces que os pacientes com SQTL2 e uma mutação fora da região do poro.
Em 2012, já é possível dizer que os testes genéticos para SQTL satisfazem amplamente a tríade, em termos de impacto diagnóstico (caso índice e familiares), prognóstico (no nível de genes e dentro de genótipos específicos) e terapêutico (abordagem diferencial de pacientes com SQTLs de acordo com a confirmação genotípica). Do ponto de vista da abordagem clínica e das possibilidades de estratificação de risco adicional, qualquer paciente com suspeita de SQTL deveria ter a opção de uma avaliação genética, mesmo nos casos em que o fenótipo clínico e eletrocardiográfico não deixem dúvidas quanto ao diagnóstico.
DISPLASIA/MIOCARDIOPATIA ARRITMOGÊNICA VENTRICULAR DIREITA (D/MAVD): A GENÉTICA COMO CRITÉRIO MAIOR DE DIAGNÓSTICO
D/MAVD é uma doença genética caracterizada por anormalidades estruturais e funcionais do ventrículo direito principalmente, com substituição progressiva do miocárdio por tecido adiposo e fibroso, decorrente de alterações do processo apoptótico. Afeta seis em cada 10 mil indivíduos. Suas manifestações clínicas podem variar entre a falta de sintomas e a presença de arritmias ventriculares e insuficiência cardíaca direita ou biventricular. Além disso, é uma das principais causas de morte súbita em adultos jovens, especialmente nos atletas. Esta é uma doença em que a genética deposita um papel muito importante, sendo que 50% dos casos pode acometer diferentes membros de uma família (agregação familiar).
O diagnóstico de D/MAVD se faz de acordo com critérios (maiores e menores) recentemente modificados em 2010 (publicados simultaneamente em Circulation e o European Heart Journal). Mutações nos genes desmossômicos têm sido reportadas em aproximadamente 50% dos pacientes diagnosticados com esta doença. Como consequência, os novos critérios diagnósticos incluem a presença de uma mutação patogênica como um critério diagnóstico maior.
É importante ressaltar que em até 7% dos casos com D/MAVD, duas ou três mutações são identificadas no mesmo paciente. Estes casos apresentam fenótipos mais severos (morte súbita, ataque cardíaco precoce, etc.).
A presença de uma mutação em um gene desmossômico não somente auxilia com o diagnóstico do paciente índice, mas também permite o rastreamento rápido dos membros da família. D/MAVD é herdada de uma maneira autossômica dominante, ou seja, se um indivíduo está afetado, seus filhos têm 50% de probabilidade de terem herdado o gene mutado e serem também afetados. Os familiares nos quais uma mutação causal é detectada devem ser monitorados, uma vez que a doença é progressiva e os sintomas podem aparecer ao longo dos anos. Por outro lado, a não detecção da mutação em um familiar permite um diagnóstico de certeza da condição de não portador e o familiar pode ser liberado da monitoração.
O DESAFIO DO DIAGNÓSTICO GENÉTICO NA MIOCARDIOPATIA DILATADA
A Miocardiopatia Dilatada (MCD) é caracterizada pela presença de dilatação e disfunção sistólica ventricular esquerda em ausência de condições de sobrecarga (hipertensão, doença valvular) ou alteração coronariana suficiente para causar deficit global da função sistólica. Afeta aproximadamente um em cada três mil indivíduos e representa a terceira causa mais comum de falha cardíaca, sendo uma causa importante de transplante cardíaco. Embora diferentes injúrias ao miocárdio possam causar um fenótipo de cardiomiopatia dilatada, o termo MCD aqui empregado identifica a doença de causa genética. Até 50% dos casos de MCD têm apresentação familiar e causa genética.
A MCD é resultante de mutações raras, principalmente missense, ou variantes de inserção/deleção. Até o presente, estas mutações foram encontradas em cerca de 30 genes diferentes, relacionados com proteínas do citoesqueleto, miofilamentos do sarcômero, uniões intercelulares, envelope nuclear, canais iônicos e proteínas mitocondriais. No entanto, nenhuma destas mutações em genes específicos representa mais de 5% da DCM familiar. O modo predominante de herança é autossômico dominante, embora possam existir, menos frequentemente, formas recessivas ligadas ao cromossomo X e de herança mitocondrial.
A sensibilidade dos testes genéticos é estimada em 15-25%. Isto varia em relação ao número de genes que são testados e às características fenotípicas da pessoa testada. Para aqueles indivíduos com doença de condução, creatina quinase elevada e familiares afetados, os testes genéticos parecem ser mais produtivos. De qualquer maneira, essa doença exige o trabalho multidisciplinar do clínico em conjunto com o laboratório para viabilizar a avaliação genética face à grande heterogeneidade clínica e multiplicidade de alvos moleculares possíveis. A figura 1, na próxima página, mostra uma proposta de abordagem molecular das MCDs de acordo com características clínicas, tendo em conta principalmente os defeitos de condução associados.
A PERSPECTIVA FAMILIAR É ESSENCIAL NA AVALIAÇÃO CLÍNICA
Nestas doenças, a utilidade clínica do foco no paciente individual é superada pela informação fornecida pela visão de que o indivíduo com suspeita de DCH faz parte de uma família. A informação familiar é de extrema importância para orientar os algoritmos diagnósticos do paciente, e essa mesma família será beneficiada posteriormente com a possibilidade de triagem, aconselhamento genético e monitoração clínica para prevenir complicações sérias. O diagnóstico de um paciente index ou índice deve acionar a triagem clínica dos parentes. O principal objetivo deste rastreamento é identificar outros membros da família em risco para complicações relacionadas à doença, incluindo a morte súbita. Mas o estudo da família também é útil para realizar o diagnóstico correto e a estratificação de risco do paciente índice.