Atualização no diagnóstico do Diabetes mellitus
Novas perspectivas A resistência insulínica e o diabetes monogênico Mody
Dra. Paula Bruna Araujo e Dra. Yolanda Schrank*
Mais de 400 milhões de pessoas em todo o mundo apresentam Diabetes Mellitus (DM). Um levantamento da Organização Mundial de Saúde (OMS), realizado em 2012, mostrou que o DM foi a principal causa direta de morte de 1,5 milhões de indivíduos e que a glicemia elevada contribuiu para a mortalidade em 2,2 milhões de pessoas.
Para o futuro, prevê-se que essa doença venha a ser a sétima principal causa de morte no ano de 2030. Apesar de o DM tipo 2 (DM2) ser o mais frequente e ocorrer principalmente em adultos, nas últimas décadas têm-se relatado aumento de DM2 também em crianças e adolescentes; igualmente o diabetes tipo 1 (DM1) tem também ocorrido nessa população, especialmente em crianças mais jovens; ambos têm se tornado um novo problema clínico dentro da prática pediátrica. Medidas que propiciem o diagnóstico precoce do DM em qualquer faixa etária e a caracterização do tipo de DM apresentado pelo indivíduo contribuem para que o médico possa tomar a conduta terapêutica adequada.
Parte 1 – Diabetes e autoimunidade: Da resistência insulínica ao diabetes tipo 1
NOVOS MARCADORES NO DIAGNÓSTICO DO DIABETES TIPO 1 AUTOIMUNE
O diabetes tipo 1 (DM1) consiste em doença crônica caracterizada pela destruição autoimune das células beta-pancreáticas. Os fatores imunogenéticos e ambientais associados ao início e à progressão da doença não estão totalmente esclarecidos.
Os autoanticorpos podem estar presentes anos antes do desenvolvimento da doença clínica, podendo, dessa forma, ser utilizados na identificação de indivíduos com maior risco de diabetes. A progressão da doença, por sua vez, está associada à presença de múltiplos autoanticorpos.
Depois da descrição dos primeiros anticorpos anti-ilhotas pancreáticas (ICAs), foram descritos vários outros autoanticorpos relevantes como anticorpos contra a insulina (IAA), a descarboxilase do ácido glutâmico (GAD), a proteína tirosina-fosfatase IA2 (IA-2) e, mais recentemente, o autoanticorpo transportador do zinco (ZnT8) (quadro 1). Considera-se, atualmente, portanto, cinco os autoanticorpos principais relacionados com o DM1.

ANTICORPO CONTRA O TRANSPORTADOR DO ZINCO (ZNT8)
O Znt8 consiste em proteína produzida pelo gene SCL30A8, localizada na membrana dos grânulos secretores de insulina das células beta. A sua função é transportar o zinco do citoplasma para o interior das vesículas. O zinco é essencial à apropriada maturação e ao armazenamento da insulina.
Os autoanticorpos para o Znt8 representam importante e promissor marcador do DM1, uma vez que mostraram prevalência de 60% a 80% em pacientes recém-diagnosticados com a doença, dos quais 14% a 26% não expressaram nenhum outro autoanticorpo.
Estudo de Kawasaki e col. tem mostrado, ainda, que a prevalência do anticorpo é maior (~58%) em pacientes com início agudo da doença e me- nor (~20%) em pacientes com início mais insidioso. Esse mesmo estudo mostrou prevalência inversa do anti-Znt8 com a idade de início do diabetes, sendo maior (~70%) em pacientes com menos de 10 anos.
Além disso, o anti-Znt8 se mostrou muito útil na previsão de DM1 em parentes de primeiro grau de pacientes com a doença.
Concluímos que incluir a dosagem do anti-Znt8 na avaliação inicial dos pacientes, com possível DM1, oferece um grande diferencial, aumentando a sensibilidade da detecção de autoimunidade para 98%.
A determinação do anti-Znt8 é realizada por enzimaimunoensaio, não sendo necessário jejum para a sua coleta.
DIAGNÓSTICO DA SÍNDROME DE RESISTÊNCIA INSULÍNICA AUTOIMUNE
A resistência insulínica autoimune consiste em causa rara da resistência insulínica, destacando-se as três causas descritas a seguir:
- Resistência insulínica secundária à presença de anticorpos anti-insulina, mais comumente descrita antes do advento das insulinas humanas;
- Síndrome de resistência insulínica do tipo A, na qual o defeito é do receptor de insulina e da cascata de sinalização pós-receptor;
- Síndrome de resistência insulínica do tipo B, que ocorre pelo desenvolvimento de imunoglobulinas policlonais (IgG) contra o receptor da insulina.
A síndrome de resistência insulínica do TIPO B (SRI-B) consiste em rara desordem de prevalência ainda desconhecida, causada pela presença de autoanticorpos policlonais contra epítopos específicos do receptor de insulina. A desordem é mais frequentemente descrita em mulheres afro-americanas, entre 30 e 50 anos, e apresenta alta mortalidade. A maioria dos pacientes desenvolve a síndrome no contexto de outra doença autoimune, sendo a associação com o lúpus eritematoso sistêmico mais frequentemente descrita. Nessa situação, usualmente o lúpus antecede o aparecimento do anticorpo antirreceptor de insulina. Dentre as demais doenças relacionadas à SRI-B, destacamos: tireoidite de Hashimoto, esclerose progressiva sistêmica, síndrome de Sjögren e trombocitopenia autoimune.
A doença pode cursar com um amplo espectro de anormalidades no metabolismo da glicose, variando de resistência insulínica extrema à hipoglicemia grave. Em estudo interessante com 24 pacientes portadores da síndrome, Arioglu et al observaram que a maioria, cerca de 80% dos pacientes, abriu o quadro com hiperglicemia e os demais apresentaram hipoglicemia como primeira manifestação da doença. Dentre os pacientes incialmente hiperglicêmicos, 15% apresentaram também episódios de hipoglicemia grave.
Os achados mais característicos da síndrome são acantose nigricans e hiperandrogenismo, achados estes comuns às demais síndromes de resistência insulínica. Destaca-se, entretanto, como característica da síndrome, a presença da acantose também em região periocular.
Tipicamente, hiperglicemia refratária e resistên- cia insulínica extrema em paciente portador de doença autoimune são os achados cardinais da SRI-B, mas, como descrito acima, a hipoglicemia também pode estar presente e consiste na principal causa da alta mortalidade da doença.
O diagnóstico laboratorial é realizado por meio da determinação dos anticorpos antirreceptores de insulina. Interessantemente, a doença pode remitir espontaneamente em cerca de um terço dos pacientes. O tratamento não está bem estabelecido, entretanto, recentes avanços terapêuticos, que consistem na combinação de imunomoduladores, têm resultado em significativa melhora na mortalidade e em períodos maiores de remissão.
Em resumo, apesar de ser uma doença rara, em vigência da tríade representada por hiperglicemia de difícil controle, resistência insulínica extrema e doença autoimune, a determinação do anticorpo antirreceptor de insulina é mandatória.
Vale lembrar que a pesquisa do anticorpo também deve fazer parte da rotina de investigação de hipoglicemia, fazendo a SRI-B inclusive diagnóstico diferencial com insulinoma, uma vez que ambas as patologias cursam com aumento da insulina. Enquanto no insulinoma esse aumento é decorrente da maior produção, na SRI-B a presença do anticorpo antirreceptor de insulina impede a sua degradação, resultando em diminuição do clearance da insulina.
AUTOANTICORPOS ANTIRRECEPTORES DE INSULINA
Os autoanticorpos antirreceptores de insulina são policlonais, com predomínio da imunoglobulina da classe G. A concentração de receptores de insulina está inversamente relacionada aos níveis crônicos de insulina aos quais estão expostos.
Estudos in vivo e in vitro têm mostrado que até 75% dos receptores podem ser perdidos na presença de insulina, o que parece ser consequência de uma maior degradação dos receptores. Anticorpos antirreceptores de insulina apresentam efeito insulinomimético em promover down regulation destes receptores, resultando em importante resistência insulínica e grave hiperglicemia. As alterações metabólicas descritas são decorrentes, portanto, tanto do bloqueio da ligação da insulina ao seu receptor como também da modulação do número de receptores.
Além de ação “bloqueadora”, podemos encontrar num mesmo paciente também ação “estimuladora” desses mesmos anticorpos, de forma que esses pacientes podem apresentar tanto hiperglicemia como hipoglicemia grave. Estudos sugerem que em indivíduos com baixos títulos do anticorpo, o efeito estimulador do receptor predominaria, enquanto em pacientes com títulos mais altos, predominaria o efeito bloqueador.
A determinação do anticorpo antirreceptor de insulina é realizada por radioimunoensaio e é recomendado jejum de 4 horas para coleta da amostra de sangue para a análise.
Concluindo, os diversos mecanismos fisiopatológicos envolvidos na ocorrência do DM1 e do DM2 justificam que um diagnóstico preciso seja realizado, a fim de proporcionar a diretriz terapêutica adequada. O uso clínico dos novos anticorpos – contra o transportador do zinco e contra o receptor de insulina – aliados aos demais exames já utilizados, podem contribuir para o diagnóstico do DM e o seu tratamento.
Parte 2 – Diabetes monogênico: Do diagnóstico clínico à confirmação molecular
O Diabetes mellitus (DM) do tipo Mody (Matu- rity-Onset Diabetes of the Young) refere-se a um grupo heterogêneo de desordens monogênicas com herança autossômica dominante, caracterizadas pela disfunção da célula ß pancreática, comprometendo a secreção de insulina. Caracteriza-se por um DM de intensidade moderada, tipicamente diagnosticado antes dos 25 anos, em geral durante a infância e a adolescência. É frequentemente confundido com DM tipo 1 e DM tipo 2, apresentando uma prevalência estimada de 1% a 2% dentre to- dos os casos de diabetes.
Adistinção do Mody para o DM tipo 1 e o tipo 2 é de grande importância, uma vez que o tratamento otimizado de cada um dos casos é diferente. Além disso, parentes de primeiro grau possuem uma chance de 50% de herdar a mesma mutação, conferindo um risco maior que 95% de desenvolvimento de diabetes ao longo da vida. Assim, o Mody é uma forma não insulino-dependente de diabetes que é definida geneticamente por mutações em diferentes genes, sendo que sua base genética só foi reconhecida na década de 1990.
APRESENTAÇÃO CLÍNICA DO MODY
Pacientes com Mody costumam apresentar as seguintes características:
1. Forte história familiar de diabetes (de qualquer tipo). O diagnóstico de hiperglicemia comumente acontece antes dos 25 anos em um ou mais familiares;
2. Padrão de herança autossômica dominante, com transmissão vertical de diabetes para, pelo menos, três gerações, e fenótipo similar dividi- do pelos membros da família acometidos;
3. Ausência de terapia com insulina por pelo me- nos cinco anos após o diagnóstico de diabetes ou a presença de níveis significativos de peptídeo C, mesmo em um paciente em tratamento com insulina;
4. Níveis de insulina costumam estar em níveis normais, apesar de inapropriadamente baixos para o grau de hiperglicemia, sugerindo defeito primário da função da célula ß, além de ausência de marcadores de autoimunidade contra as células ß;
5. Ausência de estigmas de resistência insulínica. Obesidade e sobrepeso estão raramente associados aos pacientes que possuem diabetes do tipo Mody.
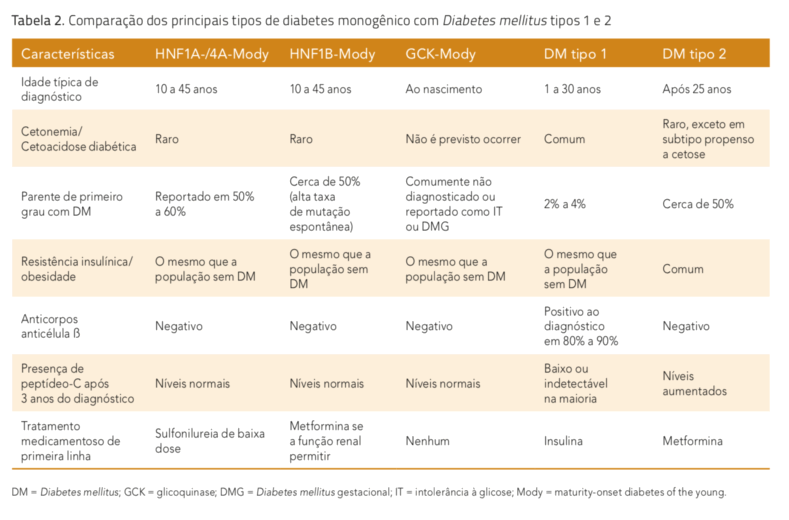
O reconhecimento do Mody pode ser um desafio pela sua baixa prevalência relativa e superposição de características clínicas com os outros tipos de diabetes. O subtipo genético específico de Mody irá determinar a apresentação clínica, o prognóstico e a resposta ao tratamento. Mutações heterozigotas ou deleções parciais ou totais em pelo menos seis genes são a causa da maioria dos casos de Mody, o que permite uma subclassificação dos subtipos de Mody (tabela 1). Mutações dos genes GCK (Mody 2) e HN- F1A (Mody 3) são as causas mais frequentes de Mody em todas as populações estudadas, corresponden- do a aproximadamente 70% de todos os casos.

MODY 2 POR MUTAÇÕES DA ENZIMA GLICOQUINASE
A enzima glicoquinase (GCK) é a enzima de fosforilação da glicose predominante nas células do parênquima hepático e nas células ß das ilhotas pancreáticas, e ambos os tipos celulares agem como um sensor aos níveis de concentração da glicose no sangue. Ela possui relativa baixa afinidade pela glicose e não é inibida por concentrações micromolares (fisiológicas) de glicose-6-fosfato, mas sim por uma proteína regulatória que transduz o efeito da frutose-6-fosfato e da frutose-1-fosfato. Mais de 600 diferentes mutações no GCK estão associadas ao Mody 2.
Estudos de propriedades cinéticas da GCK mutante mostram que há uma redução relativa da atividade enzimática, levando à redução do fluxo glicolítico em células ß pancreáticas. Dessa forma, ocorre um defeito no sensor de glicose, levando a um quadro de hiperglicemia de jejum moderada (100 a 145 mg/dL), que desencadeia a secreção de insulina. A redução do armazenamento de glicogênio hepático e o aumento da gliconeogênese hepática após refeições usuais contribuem para a hiperglicemia pós-prandial nesses pacientes. Além disso, a hemoglobina glicada (HbA1c) pode estar normal e não costuma ultrapassar 8%.
Mody 2 representa 20% a 30% de todos os casos de diabetes monogênico. Apesar de a hiperglicemia poder estar presente desde o nascimento, a maioria dos casos é detectada mais tardiamente durante rastreio incidental de glicose. Evidências de estudos observacionais mostram que esses pacientes não desenvolvem complicações microvasculares relacionadas ao diabetes, portanto, a confirmação de mutação do GCK permitiria a parada da terapia de redução da glicose e denota um prognóstico favorável sem risco de progressão do diabetes.
Durante a gravidez, mulheres com mutação GCK têm 50% de chance de ter um bebê sem mutação GCK, e, nesse caso, existe um risco aumentado de macrossomia e suas consequências obstétricas. Assim, o tratamento da mãe, com insulina, é indicado. Por outro lado, se a mãe carrega um bebê com uma mutação GCK, não é necessário tratamento. O monitoramento ultrassonográfico do tamanho fetal é recomendado para auxiliar na decisão médica de diminuir ou não a glicemia materna com insulina durante a gravidez.
OUTRAS FORMAS DE MODY DEVIDO A MUTAÇÕES EM FATORES DE TRANSCRIÇÃO EXPRESSOS PELAS CÉLULAS ß
A família dos fatores nucleares de hepatócitos (HNF) são fatores de transcrição necessários para o correto funcionamento das células ß pancreáticas. Três sub-tipos de mutação de HNF estão mais comumente associados ao diabetes monogênico: HNF1A, HNF4A e HNF1B, sendo a mutação HNF1A a mais comum. Além disso, mutações nos genes dos fatores de transcrição IPF1 e NeuroD1, também levam à disfunção da célula ß. Pacientes com Mody 3 (HNF1A) e Mody 1 (HNF4A) costumam ser clinicamente indistinguíveis, sendo tipicamente normoglicêmicos durante a infância, mas apresentam um defeito progressivo na secreção de insulina, com o diabetes sendo diagnosticado comumente da segunda à quinta década de vida. A manifestação clínica inicial costuma ser hiperglicemia pós-prandial com normoglicemia de jejum, progredindo para diabetes franco com o passar do tempo.
Mody 1 (HNF4A) também está associado ao peso aumentado ao nascer e à tendência à hipoglicemia neonatal, o que parece refletir a hiperinsulinemia intrauterina (efeito paradoxal, já que após o nascimento poderá ocorrer a redução da secreção da insulina).
Mais de 200 diferentes mutações do HNF1A já foram descritas, enquanto mutações do HNF4A são mais ra- ras. Mody 3 (HNF1A) e Mody 1 (HNF4A) frequentemente apresentam repostas excelentes e duradouras à baixa dose de terapia com sulfonilureias. No entanto, em contraste com Mody 2 (GCK), Mody 3 (HNF1A) é uma forma mais severa de diabetes, comumente evoluindo para dependência de insulina (tabela 2). Pacientes com Mody 3 e Mody 1 apresentam risco de complicações micro e macrovasculares comparável ao do DM tipo 1 e tipo 2, se correlacionando com o controle glicêmico.
Mutações no HNF1 ß são responsáveis pelo início precoce de diabetes, consistente com diagnóstico de Mody em diversas famílias e por doenças graves dos rins, incluindo malformações, que podem aparecer antes da alteração da glicemia. O fenótipo mais comum no Mody 5 (HNF1ß) é a presença de cistos renais e/ou anormalidades histológicas como meganéfrons. A associação de cistos renais com diabetes tem sido reconhecida como uma síndrome clínica chamada RCAD (Renal Cysts and Diabetes Syndrome).
Hnf-1ß desempenha um papel importante no desenvolvimento do rim e na diferenciação dos néfrons e é também um regulador crítico de uma rede transcricional que controla a especificação, o crescimento e a diferenciação do pâncreas embrionário. Outras alterações clínicas também podem ser observadas como malformações do trato urogenital, alteração dos exames de função hepática, atrofia e insuficiência exócrina pancreática, gota e hiperuricemia. Terapia com insulina costuma ser necessária em pacientes com Mody 5 (HNF1ß), de- vido à atrofia pancreática.
Dois outros genes codificantes de fatores de transcrição, IPF1 e NeuroD1, possuem um papel importante no desenvolvimento do pâncreas endócrino, mas representam causas raras de Mody. O IPF1 age como um regulador transcricional maior de genes específicos do pâncreas endócrino em adultos, como o gene da pré-proinsulina, genes GLUT2 e GCK em células ß e gene da somatostatina em células.
Pacientes com Mody 4 (IPF1) possuem fenótipo que varia desde intolerância à glicose até o diabetes não insulino-dependente. Já o fator de transcrição NeuroD1 tem papel importante no desenvolvimento do pâncreas e do sistema nervoso e regula a expressão do gene da insulina por meio da ligação à sua região promotora. Assim, a ligação deficiente da NeuroD1 ou a ligação de um polipeptídeo inativo à região promotora-alvo do gene da insulina nas ilhotas pancreáticas pode levar ao desenvolvimento de diabetes precoce em humanos (Mody 6).
MARCADORES BIOQUÍMICOS QUE AJUDAM NO DIAGNÓSTICO DO DIABETES MONOGÊNICO
O peptídeo C, um remanescente da clivagem da proinsulina, é secretado pela célula ß em concentrações equivalentes à liberação de insulina, servindo como marcador de produção endógena de insulina. Dessa forma, sua medida é particularmente útil na diferenciação de DM tipo 1 do diabetes monogênico, já que usualmente espera-se níveis indetectáveis de peptídeo C após cinco anos de duração de DM tipo 1. Os autoanticorpos contra as células ß também podem ser usados para diferenciar o DM tipo 1 do diabetes monogênico, já que 98% dos pacientes com diagnóstico recente de DM tipo 1 irão apresentar, pelo menos, um dos tipos de autoanticorpos detectável (anti-GAD, IA-2, IAA, ZnT8A), enquanto a prevalência desses anticorpos em pacientes com HNF1A, GCK ou HNF4A é de apenas 1% (comparável a casos controle).
A dosagem de proteína C reativa (PCR) pode ser particularmente útil na diferenciação do Mody 3 (HNF1A) para as outras formas de diabetes, uma vez que o Hnf-1A se liga na região promotora do gene codificante da PCR. Consequentemente, níveis marcadamente reduzidos de PCR são vistos no diabetes monogênico causado por mutação no HNF1A, comparado com outras formas de diabetes.
Níveis normais ou elevados de colesterol HDL indicam que a resistência insulínica não é um componente importante da doença, tornando o diagnóstico de DM tipo 2 menos provável (caracteristicamente associado a níveis reduzidos de HDL). Assim, níveis de HDL acima de 43 mg/dL apresentam um moderado poder discriminatório entre Mody 3 (HN- F1A) e pacientes com DM tipo 2.
DIAGNÓSTICO MOLECULAR DO DIABETES MONOGÊNICO
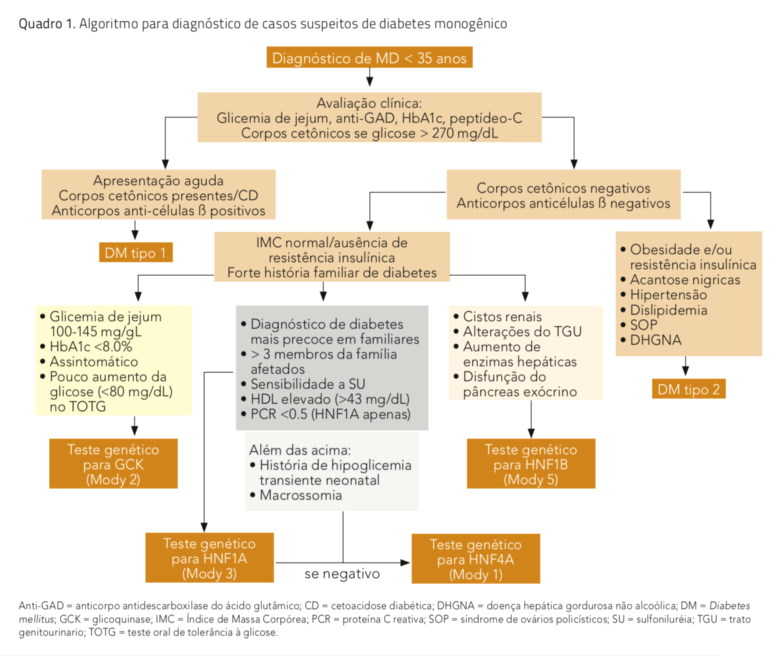
O teste diagnóstico atualmente utilizado para Mody é o sequenciamento genético pelo método de Sanger, considerado o padrão ouro para a detecção de mutações nas regiões codificantes dos genes envolvidos. A dosagem dos genes por kits comerciais de MLPA (Multiplex Ligation-dependent Probe Amplification) é necessária para a identificação de deleção parcial ou total de um gene, responsável por 1% a 3% das mutações. A testagem genética é normalmente restrita a um subgrupo de genes, de acordo com o fenótipo do paciente (quadro 1).
CONCLUSÃO
A identificação de pacientes com Mody continua sendo um desafio na prática clínica, apesar da disponibilidade de testes genéticos moleculares específicos. Por ser uma causa rara de diabetes, os médicos precisam estar cientes dessa etiologia e não hesitar em questionar um diagnóstico prévio de outro tipo de diabetes se o fenótipo for sugestivo de Mody. Assim, a confirmação do diabetes monogênico é importante, já que possui etiologias moleculares distintas, explicando uma parte substancial da heterogeneidade clínica do Mody, com grandes diferenças no curso clínico da doença e nas estratégias de tratamento a longo prazo.
REFERÊNCIAS
*Dra. Paula Bruna Araujo
Médica endocrinologista da DASA; Mestre em medicina (área de concentração
em endocrinologia) pela Universidade Federal do Rio de Janeiro; Clinical Fellowship em endocrinologia oncológica pela University of Toronto.
*Dra. Yolanda Schrank
Médica endocrinologista do corpo clínico da DASA-RJ; Médica endocrinologista do corpo clínico do HFB; Título de especialista em endocrinologia e metabologia pela SBEM/AMB; Mestre em endocrinologia pela PUC-RJ.